
Authors: Colomban de Vargas1,2,†,*, Stéphane Audic1,2,†, Nicolas Henry1,2,†, Johan Decelle1,2,†, Frédéric Mahé3,1,2,†, Ramiro Logares4, Enrique Lara5, Cédric Berney1,2, Noan Le Bescot1,2, Ian Probert6,7, Margaux Carmichael1,2,8, Julie Poulain9, Sarah Romac1,2, Sébastien Colin1,2,8, Jean-Marc Aury9, Lucie Bittner10,11,8,1,2, Samuel Chaffron12,13,14, Micah Dunthorn3, Stefan Engelen9, Olga Flegontova15,16, Lionel Guidi17,18, Aleš Horák15,16, Olivier Jaillon9,19,20, Gipsi Lima-Mendez12,13,14, Julius Lukeš15,16,21, Shruti Malviya8, Raphael Morard22,1,2, Matthieu Mulot5, Eleonora Scalco23, Raffaele Siano24, Flora Vincent13,8, Adriana Zingone23, Céline Dimier1,2,8, Marc Picheral17,18, Sarah Searson17,18, Stefanie Kandels-Lewis25,26, Tara Oceans Coordinators‡, Silvia G. Acinas4, Peer Bork25,27, Chris Bowler8, Gabriel Gorsky17,18, Nigel Grimsley28,29, Pascal Hingamp30, Daniele Iudicone23, Fabrice Not1,2, Hiroyuki Ogata31, Stephane Pesant32,22, Jeroen Raes12,13,14, Mike Sieracki33,34, Sabrina Speich35,36, Lars Stemmann17,18, Shinichi Sunagawa25, Jean Weissenbach9,19,20, Patrick Wincker9,19,20*, Eric Karsenti26,8,*
Affiliations: 1 CNRS, UMR 7144, Station Biologique de Roscoff, Place Georges Teissier, 29680 Roscoff, France. 2 Sorbonne Universités, UPMC Univ Paris 06, UMR 7144, Station Biologique de Roscoff, Place Georges Teissier, 29680 Roscoff, France. 3 Department of Ecology, University of Kaiserslautern, Erwin-Schroedinger Street, 67663 Kaiserslautern, Germany. 4 Department of Marine Biology and Oceanography, Institute of Marine Science (ICM)-CSIC, Pg. Marítim de la Barceloneta 37-49, Barcelona E08003, Spain. 5 Laboratory of Soil Biology, University of Neuchâtel, Rue Emile-Argand 11, 2000 Neuchâtel, Switzerland. 6 CNRS, FR2424, Roscoff Culture Collection, Station Biologique de Roscoff, Place Georges Teissier, 29680 Roscoff, France. 7 Sorbonne Universités, UPMC Univ Paris 06, FR 2424, Roscoff Culture Collection, Station Biologique de Roscoff, Place Georges Teissier, 29680 Roscoff, France. 8 Ecole Normale Supérieure, Institut de Biologie de l’ENS (IBENS), and Inserm U1024, and CNRS UMR 8197, Paris, F-75005 France 9 CEA, Institut de Génomique, GENOSCOPE, 2 rue Gaston Crémieux, 91000 Evry, France. 10 CNRS FR3631, Institut de Biologie Paris-Seine, F-75005, Paris, France. 11 Sorbonne Universités, UPMC Univ Paris 06, Institut de Biologie Paris-Seine (IBPS), F-75005, Paris, France. 12 Department of Microbiology and Immunology, Rega Institute, KU Leuven, Herestraat 49, 3000 Leuven, Belgium. 13 Center for the Biology of Disease, VIB, Herestraat 49, 3000 Leuven, Belgium. 14 Department of Applied Biological Sciences, Vrije Universiteit Brussel, Pleinlaan 2, 1050 Brussels, Belgium. 15 Institute of Parasitology, Biology Centre, Czech Academy of Sciences, Branišovská 31, 37005 České Budějovice, Czech Republic. 16 Faculty of Science, University of South Bohemia, Branišovská 31, 37005 České Budějovice, Czech Republic. 17 CNRS, UMR 7093, LOV, Observatoire océanologique, F-06230, Villefranche-sur-mer, France. 18 Sorbonne Universités, UPMC Univ Paris 06, UMR 7093, LOV, Observatoire Océanologique, F-06230, Villefranche-sur-mer, France. 19 CNRS, UMR 8030, CP5706, Evry, France. 20 Université d'Evry, UMR 8030, CP5706, Evry, France. 21 Canadian Institute for Advanced Research, 180 Dundas Street West, Suite 1400, Toronto ON M5G 1Z8, Canada. 22 MARUM, Center for Marine Environmental Sciences, University of Bremen, 28359 Bremen, Germany. 23 Stazione Zoologica Anton Dohrn, Villa Comunale, 80121 Naples, Italy. 24 Ifremer, Centre de Brest, DYNECO/Pelagos CS 10070, 29280 Plouzané, France. 25 Structural and Computational Biology, European Molecular Biology Laboratory, Meyerhofstr. 1, 69117 Heidelberg, Germany. 26 Directors’ Research, European Molecular Biology Laboratory, Meyerhofstr. 1, 69117 Heidelberg, Germany. 27 Max-Delbrück-Centre for Molecular Medicine, 13092 Berlin, Germany. 28 CNRS UMR 7232, BIOM, Avenue du Fontaulé, 66650 Banyuls-sur-Mer, France. 29 Sorbonne Universités Paris 06, OOB UPMC, Avenue du Fontaulé, 66650 Banyuls-sur-Mer, France. 30 Aix Marseille Université, CNRS IGS UMR 7256, 13288 Marseille, France. 31 Institute for Chemical Research, Kyoto University, Gokasho, Uji, Kyoto, 611-0011, Japan. 32 PANGAEA, Data Publisher for Earth and Environmental Science, University of Bremen, Bremen, Germany. 33 Bigelow Laboratory for Ocean Sciences, East Boothbay, ME, USA. 34 National Science Foundation, Arlington, VA, USA. 35 Department of Geosciences, Laboratoire de Météorologie Dynamique (LMD), Ecole Normale Supérieure, 24 rue Lhomond, 75231 Paris Cedex 05, France. 36 Laboratoire de Physique des Océans, UBO-IUEM, Place Copernic, 29820 Plouzané, France.
The biological and physico-chemical samples were collected during the circumglobal expedition Tara Oceans (fig. W1), which sampled entire planktonic communities from phages to small metazoans across 11 organismal size-fractions and >6 orders of size magnitude. For eukaryotic plankton genetics, 4 size-fractionated communities were usually obtained from two depths in the photic zone (subsurface and Deep-Chlorophyll Maximum (DCM)), with appropriate gears: a low-shear and non-intrusive industrial peristaltic pump for the piconano-plankton (0.8-5 μm) and plankton nets for the nano-, micro-, and meso- plankton (respectively, 5-20μm, 20-180 μm and 180-2000 μm). The volumes of filtered seawater were scaled according to known organismal concentrations within each size fraction, from 0.1 m3 (100 L) for the most concentrated pico-plankton to 148±136m3 for the most-dilute meso-plankton (database W1), in order to get near-exhaustive recovery of total eukaryotic biodiversity in each sample. Whole plankton communities were subsequently filtered on polycarbonate membranes, rapidly flash-frozen and preserved in liquid nitrogen on board Tara, and stored cryopreserved in the laboratory until nucleic acids extraction and sequencing. A detailed description of all Tara Oceans field sampling strategy and protocols is available in (Pesant et al. n.d.) and database W1 lists all analyzed samples and their metadata.
Cryopreserved plankton polycarbonate membranes were cryo-crushed with 10 knocks per second for 1 minute using a FreezerMill 6700 (Fisher Scientific), yielding approximately 1 g of material per membrane. Total DNA (and RNA) were extracted simultaneously from each membrane using the NucleoSpin®RNA L kit combined with DNA Elution buffer kit (Macherey-Nagel). Extracted nucleic acids were resuspended in 3.6 ml RA1 lysis buffer and 36 µL β-mercaptoethanol, vortexed for 1 min (5 sec bursts), transferred to a NucleoSpin Filter L column (Macherey-Nagel) and spun for 10 min at 4,500 g. The eluate was transferred to a new tube and the nucleic acids were precipitated using 3.6 ml of 70% ethanol. Samples were loaded into a NucleoSpin RNA L column (Macherey-Nagel). The column was washed twice with DNA wash solution, and DNA was eluted in 400 µL of DNA elution buffer. RNA was treated separately. Total DNA was quantified using a Nanodrop ND-1000 Spectrophotometer (Labtech International) and quality checked on an agarose gel (1.5 %). PCR amplifications of the hyper-variable loop V9 of the 18S rRNA gene was performed with the Phusion® High-Fidelity DNA Polymerase (Finnzymes) and the forward/reverse primer-pair 1389F 5’- TTGTACACACCGCCC -3’ and 1510R 5’- CCTTCYGCAGGTTCACCTAC -3’ (Amaral-Zettler et al. 2009). The PCR mixtures (25 µL final volume) contained 5 ng of total DNA template with 0.35µM final concentration of each primer, 3% of DMSO and 2X of GC buffer Phusion Master Mix (Finnzymes). PCR amplifications (98 °C for 30 sec; 25 cycles of 10sec at 98 °C, 30 sec at 57 °C, 30sec at 72 °C; and 72°C for 10 min) of all samples were carried out with a reduced number of cycles to avoid the formation of chimeras during the plateau phase of the reaction, and in triplicate in order to smooth the intra-sample variance while obtaining sufficient amounts of amplicons for Illumina sequencing. PCR products were run on a 1.5% agarose gel to check amplicon lengths. Amplicons were then pooled and purified using the NucleoSpin® Extract II kit (Macherey-Nagel), and quantified with the Quant-iT™ PicoGreen® dsDNA kit (Invitrogen). Bridge amplification and paired-end sequencing of the amplified fragments were performed using a Genome Analyser IIx system (Illumina, San Diego, CA, USA) with chemistry version 5, and software version SCS 2.9.35.0 and RTA 1.9.35.0 for sequences produced in 2011 and 2012 and version SCS 2.10 and RTA 1.13.48 for sequences produced in 2013.
Our bioinformatics pipeline allowed filtering of high-quality V9 rDNA sequences (metabarcodes) and their clustering into operational taxonomic units (OTUs, fig. W2A). Overlapping reads were merged using a custom script based on the fastx software (http://hannonlab.cshl.edu/fastx_toolkit/index.html), the last nucleotides of the right member of a read pair being aligned to its cognate read. Paired reads were retained for downstream analyses if they contained both forward and reverse primers. For each sample, reads were then dereplicated. Reads present as a single copy and in a single sample were further filtered based on quality values from the paired fastq file, by evaluating the expected error in a 50 bp sliding window and discarding sequences with more than 1% of error in the worst quality window. The filtered reads were then checked with the chimera search module of the usearch program (version 4.2; (Edgar et al. 2011)), looking for chimeras both with respect to the V9_PR2 reference database (database W2), as well as de novo within each sample by checking for sequences that could be chimeras arising from the most abundant environmental sequences. Predicted chimeras seen only in a single sample were discarded. A summary table was compiled, including V9 rDNA metabarcodes (a barcode being defined as a unique rDNA read) occurrence and abundance across samples. This table was further reduced by requiring that every metabarcode be (i) observed in two distinct samples (one confirming the other) and (ii) present in at least three copies amongst all samples. The rationale is that if we redo the same total experiment and if occurrence of metabarcodes is Poisson distributed, then those metabarcodes with an abundance of 3 would have more than 90% of chance to still be present (93.75%). Metabarcodes were then clustered into biologically meaningful OTUs, using the ‘Swarm’ approach (Mahé et al. 2014) (fig. W2B). In most analyses dealing with biodiversity, the most abundant metabarcode of each ‘swarm’ was chosen as a representative of its OTU.
For the taxonomic assignation of all Tara Oceans metabarcodes and OTUs, we assembled a new database of reference, taxonomically recognized, Sanger-sequenced V9-rDNA barcodes, called V9_PR2 (database W2) and derived from the Protist Ribosomal Reference -PR2- database (GenBank release 191, (Guillou et al. 2013)). The V9 sequences were first extracted from the partial and complete reference 18S rDNA sequences from PR2, and the forward and reverse PCR primer sequences were removed. Because the PCR primers used in this study can potentially amplify prokaryotic taxa, all prokaryotic 16S rDNA sequences from the SILVA ‘All species living tree project’ (http://www.arb-silva.de/projects/living-tree/; release LTPs111, February 2013) were extracted, truncated to the V9 fragment, and added to V9_PR2 for accurate discrimination between eukaryotic and prokaryotic rDNA. Further modifications were made to the original structure of PR2: (i) taxonomic ranks were extended for a few eukaryotic lineages requiring finer taxonomic resolution, such as the copepods (classified as Maxillopoda/genus/species in PR2, and refined in V9_PR2 by inserting an additional ‘Order-level’ taxonomic field -Calanoida, Cyclopoida, Harpacticoida, Misophrioida, Monstrilloida or Siphonostomatoida); (ii) names were modified at various taxonomic levels. ‘Dino-Group-X’ was replaced by the more accepted ‘Marine ALVeolates’ MALV-I, II, III and IV; the new classification of ‘MAST’ (Marine Stramenopiles) ribogroups and the novel environmental clades ‘MOCH’ (Marine Ochrophytes) (Massana et al. 2014) were incorporated; an expert-curated classification of dinoflagellates was implemented; (iii) the environmental reference sequences from PR2 (typically from Sanger-sequenced environmental clones libraries) were kept only if they were different from taxonomically named sequences or clusters, forming entirely new ribotypes or clades. Furthermore, all V9_PR2 sequences were clustered and reference clusters with conflicting annotations were manually curated. The final V9_PR2 database comprising a total of 77,449 reference V9 rDNA barcodes is available as database W2. Ultimately, each Tara Oceans metabarcode (and thus OTU) was taxonomically assigned to the V9_PR2 reference database using the global alignment search strategy implemented in the ggsearch36 program (Fasta package, http://faculty.virginia.edu/wrpearson/fasta/CURRENT/). For each metabarcode, the best hit (% identity) to any reference barcode was retained. In case of equality, as much as 20 best hitting barcodes were retained and taxonomic assignment was based on the name of the last common ancestor.
Phylogenetic trees based first on all V9 rDNA reference barcodes were reconstructed for each of the 85 major eukaryotic lineages represented in Fig. 3, main paper. Group-specific V9 rDNA reference sequences were extracted from the V9_PR2 database and sequences with identical taxonomic path and nucleotide sequence were merged. Sequences were then aligned using the mafft program (Version 6.903; Katoh and Frith 2012), and neighbor-joining phylogenetic trees were reconstructed using the ninja program (http://nimbletwist.com/software/ninjaP). These reference trees were used to check visually the taxonomic resolution and coherence of V9 rDNA barcodes within each of the 85 major eukaryotic lineages considered. Tara Oceans V9 rDNA metabarcodes were subsequently added to the phylogenetic analyses with the following steps: (i) representative sequences of each Tara Oceans OTU were extracted, filtered on total abundance of OTUs if necessary to collect at most 1,000 V9 rDNA metabarcodes; (ii) the metabarcodes were then added to the original reference multiple alignment based on V9_PR2 barcodes (option -add in mafft), (iii) a final tree containing both reference and Tara Oceans sequences was reconstructed with ninja. For each of the 85 eukaryotic lineages represented in Fig.3, trees based on reference barcodes and trees based on reference + Tara Oceans (meta)barcodes are available in database W9. Example trees are given for the Bacillariophyta and Acantharea (fig. W7). All trees were further used to assess the phylogenetic novelty brought by Tara Oceans metabarcodes to total protistan rDNA knowledge. Total branch length was computed and compared between the reference tree (containing exclusively reference V9 rDNA sequences) and the tree containing both reference and Tara Oceans sequences. Tree-length increase (%) was used as a proxy of phylogenetic novelty for each major eukaryotic lineage (fig. W6A) or super-group (fig. W6B).
Differences in community composition amongst samples were computed using Bray-Curtis dissimilarity after a double normalization procedure. Starting from a table of the counts of each Tara Oceans V9 rDNA OTU (or metabarcode) in every sample (and considering only sequences assigned to eukaryotes), the abundance of each OTU was normalized to the maximum value that it reaches in any sample, and then expressed for each sample as a fraction of the total OTU abundance (thus summing to 1). The Bray-Curtis dissimilarity matrix was then computed from this normalized matrix and Non-Linear-Multidimensional Scaling was applied to it, using the metaMDS command from the 'vegan' R package (http://CRAN.R-project.org/package=vegan).
We are aware of the two major limitations of using nuclear rDNA (meta)barcoding systems for exploring diversity patterns in eukaryotes. Firstly, rates of rDNA substitution can be relatively slow in some eukaryotic lineages and can differ (and thus so can taxonomic resolution) between eukaryotic lineages. Even the most variable regions of 18S rDNA such as the V9 loop cannot occasionally discriminate between closely related species (Pawlowski et al. 2012). Secondly, rDNA barcodes are found in multiple, sometimes slightly different (Decelle et al. 2014; Pillet, Fontaine, and Pawlowski 2012; Santos and Kinzie 2003) copies in single eukaryotic genomes (text W2 and fig. W3). Nevertheless, the V9 rDNA barcode presents a unique combination of advantages that make it a highly versatile and exceptional tool for addressing general questions of eukaryotic diversity over holistic taxonomic and ecological scales. First, V9 is a structurally simple region with a relatively short and stable length across all eukaryotic lineages (130±4bp), and it is flanked by highly conservative sequences allowing universal-eukaryote PCR priming. This allows relatively unbiased PCR amplification of total eukaryotic rDNA diversity in environmental samples (unlike other rDNA variable regions, such as the V4, whose structural complexity and variable length can generate strong biases against entire lineages), a requisite to cover the enormous biodiversity of eukaryotic life present in oxygenic systems. Note that our metabarcode dataset contained ~5% of diverse prokaryotic sequences (Fig. 2A, main paper), which further witnesses the universality of the eukaryotic primers used (Amaral-Zettler et al. 2009). Secondly the V9 rDNA contains both stable and highly variable sequences, allowing both coherent phylogenetic placement at the class to family levels and resolution of biodiversity patterns at lower taxonomic levels, respectively. Third the number of rDNA copies per genome has been shown to correlate positively to size and/or volume of cells across a wide taxonomic range of eukaryotes, and can therefore be used as a rough proxy for the biovolume of the taxon it represents (see text W2). Last but not least, V9 is a piece of 18S rDNA which is still nowadays by far the most represented reference marker associated to described eukaryotic taxa in public databases. Thus, we propose that V9 rDNA OTUs and reads, when compared to an accurate functionally-annotated reference database, represent unique and coherent proxies for assessing patterns of functional biodiversity and biovolumes of holistic communities of eukaryotes in complex environmental systems.
rDNA gene copy numbers vary from one to hundreds of thousands in single eukaryotic genomes (Godhe et al. 2008; Gong et al. 2013; Wyngaard et al. 1995; Zhu et al. 2005), oftentimes precluding direct translation of rDNA reads into numbers of individual organisms in a given sample. However several independent studies over the last ca. 20 years have shown that the number of rDNA copies per genome correlates positively to the size and/or biovolume of the cell across a wide taxonomic and size range of eukaryotes (Godhe et al. 2008; Gong et al. 2013; Zhu et al. 2005). Here we compiled all data comparing cell length and rDNA copy numbers available in the literature (fig. W3A). Despite the different molecular techniques used to assess rDNA copy number in these studies spanning two decades, the compiled data confirm that copy number tends to increase with the size of the organism: cells smaller than 5µm generally have between 1 and 5 rDNA copies while cells larger than 200µm have between 10,000 and 200,000 copies. Although dinoflagellates are known to have very large genomes, rDNA copy numbers are not particularly out of the range but follow the overall trend (e.g., Prorocentrum sp., Peridinium sp. Amphidinium sp. etc). Some ciliates are clear outliers, which can be explained by their unique dual genome architecture and differential gene amplification patterns in their macronuclei, which can disassociate rDNA abundances from cell volume. The positive correlation between eukaryotic rDNA copy number and cell size is likely even better when using organismal volume instead of size, as shown in (Godhe et al. 2008) and below (fig. W3 B and C), suggesting that rDNA copy number is a coherent proxy for taxon-specific biovolume.
To further verify whether the molecular protocol used to generate the Tara Oceans V9 rDNA metabarcoding dataset preserves the correlation between rDNA copy number and organismal biovolume, light microscopy counts of phytoplankton taxa were performed on microplanktonic (20-180μm) samples from nine Tara Oceans stations from the Indian, Atlantic, and Southern Oceans (fig. W3, B and C). For comparison between eukaryotic supergroups (coccolithophores, diatoms, and dinoflagellates), taxa-specific counts were converted to biovolume and biomass based on cell size measurements, and counts, biovolumes and biomasses were all compared to V9 rDNA read numbers from the same samples (fig. W3B). The results confirm that V9 rDNA read numbers (in relative abundance) correlate significantly better to biovolume (r2=0.97, p-value=10-16), and cannot be used as reliable comparative proxies for taxa abundances (not significantly correlated). For instance, at Station TARA_078, coccolithophores represent ~25% of microscopy counts whereas they contribute to <1% of both V9 rDNA reads and organismal biovolumes. However, when utilized within a restricted eukaryotic lineage and organismal size-fraction, V9 rDNA read number can even be a good proxy for taxa abundance. For example, comparison of microscopy counts and V9 rDNA reads numbers within the diatoms (bacillariophyta) displayed a good match at the genus level (fig. W3C). Therefore, V9 rDNA OTUs and Reads are self-coherent, rough proxies for assessing, respectively, the biodiversity and biovolume of near-exhaustive communities of eukaryotes, and allow at the same time to zoom across the taxo-functional structure of the biodiversity, if compared to a reference database annotated with appropriate functions. All these properties were used to interpret the biocomplexity of our dataset.
Metabarcodes with ≥ 80% identity to a reference V9 rDNA barcode (ggsearch) were considered as assignable, while below this threshold the relatively small size and fast rate of substitution of the V9 rDNA loop make it virtually impossible to distinguish between eukaryotic supergroups. On the known, assignable side of the biodiversity spectrum (fig. 2A, main paper), we note the overall dominance of Alveolata in both richness and abundance, especially in the piconano- and nano-plankton, where dinoflagellates and parasitic marine alveolates (MALV) make up, respectively, ~31% and ~12% of all known eukaryotic diversity. Opisthokonta, represented essentially by metazoan OTUs, is the second most-diverse supergroup, with an increase in both richness and abundance toward larger organismal size-fractions as expected. Surprisingly, the Excavata and Rhizaria supergroups, which are largely ignored in modern plankton studies, represent ~26% of total known eukaryotic diversity, largely above Stramenopila and Archaeplastida which contain most classical phytoplankton lineages but account together for only ~8% of the known diversity. Excavata and Rhizaria display strikingly opposite patterns in terms of abundance, the Rhizaria becoming overwhelmingly abundant in larger size-fractions, mirroring metazoans. On the other, ‘unknowable’ hand, some evidence indicates that the ~35% of 'unassignable’ OTUs belonged in majority to non-referenced eukaryotic taxa. First we noted that the large majority of eukaryotic V9 fragments start with GTCG while the prokaryotic fragments start with GTCA. According to this criteria, ~78% and 61% of ‘unassignable’ reads and OTUs, respectively, are of eukaryotic origin. Two other kinds of analyses - metabarcode taxonomic assignment without thresholding and differential size-distribution spectrum between eukaryotic and prokaryotic V9 fragments - confirm independently that 65% to 58% of the reads and 48% to 47% of the OTUs belonged to unknown eukaryotic taxa.
Several well-known eukaryotic lineages are totally missing from plankton communities in the photic zone, and so are not shown in Fig. 3 (see database W6). These can be entire high-taxonomic level groups or more specific lineage(s) within wider groups that contain marine planktonic members. They can be divided into two categories: groups present in marine habitats but with an exclusively benthic lifestyle, and groups that are exclusively adapted to terrestrial environments. Examples in the first category are (i) Breviate amoeboflagellates, an ancient, independent higher-level group of eukaryotes incertae sedis probably related to Apusozoa and known from anoxic or hypoxic marine and freshwater sediments only; (ii) several Amoebozoa lineages such as the large (200 to 800 μm) amoebids, the testate arcellinids, all large reticulate species within class Variosea, and the dictyosteliid slime molds; (iii) within Stramenopila, the reticulate Synchromophyceae and the network-forming genus Labyrinthula; and (iv) several Rhizaria lineages such as the testate Gromiids, the reticulate Filoretids, some predominantly amoeboid Chlorarachnea, and the vast majority of known Foraminifera lineages. Many of these lineages correspond to large amoeboid organisms, often highly branched or reticulate, and therefore adapted to life in particulate benthic habitats exclusively. Groups missing from the photic zone plankton because they are strictly terrestrial can be free-living organisms such as land plants (Embryophyta) or lineages of exclusively freshwater or soil flagellates within groups like Apusozoa, Cryptophyta, Excavata, and Cercozoa (the hugely diversified soil order Glissomonadida and the genus Cercomonas in particular). But many of them correspond to lineages of symbionts or parasites of terrestrial plants and metazoans (especially insects and tetrapod vertebrates). Notable examples include: (i) land plant symbionts/parasites such as the glomeromycetes (endomycorrhizal Fungi), plasmodiophorids (Rhizaria), and some oomycetes (Stramenopila); (ii) most non-gregarine apicomplexan parasites (Cryptosporidium, Toxoplasma, Babesia, Plasmodium, and their relatives); (iii) about half of the lineages within the supergroup Excavata, such as oxymonads (gut symbionts in wood-eating insects), parabasalids (Trichomonas and relatives), diplomonads (Giardia and relatives) and all trypanosomes; and (iv) a few other parasites of metazoans such as Blastocystis (Stramenopila) and Entamoeba (Amoebozoa). This vast diversity of eukaryotic life missing from photic zone plankton is consistent with the hypothesis of a marine benthic origin of most eukaryotic lineages, with relatively limited numbers of lineages having adapted to a planktonic lifestyle.
Beyond the hyper-diverse eukaryotic lineages, our dataset revealed considerable phylogenetic diversity (>50 deep-branching groups) of poorly known heterotrophic organisms with important implications for marine plankton ecology. Phagotrophic nano-flagellates, which play a key role in planktonic ecosystems as major bacterial grazers, are represented mainly by Katablepharidophyta (413 OTUs) and Telonemia (240 OTUs), but their numbers are likely to increase significantly when the ecology of members of unexplored clades (i.e., MASTs, diplonemids) is investigated. Amongst osmotrophs, marine fungi are represented mainly by yeasts. Ascomycetes are very diverse (410 OTUs) and include the ubiquitous Candida, the halotolerant Hortaea, plus OTUs representing a novel marine fungal diversity affiliated to the Saccharomycetales, with great potential for industrial applications. In contrast, Basidiomycetes are represented by fewer lineages, and most sequence reads belong to Pseudozyma (>85%), followed by the ubiquitous Rhodotorula. Other important osmotrophic organisms include Labyrinthulea (322 OTUs), which can also be parasitic or mutualist. This extensive phylogenetic diversification of essentially small (<5μm) phagotrophic and osmostrophic protists could be driven by specialization on prey and/or organic molecules. However, their biodiversity is not as dramatic as in terrestrial soil systems where minute phagotrophic cercozoans (Howe et al. 2009) and osmotrophic fungi (Jones and Richards 2011) are hyper-diverse, reflecting the structural complexity of their food. On the other hand, parasite (and especially parasitoid) diversity is tremendous in our dataset, encompassing >10,000 OTUs if one takes into account only the known groups. Most of the hyper-diverse lineages of eukaryotic plankton interact with groups of parasites/parasitoids which were detected in our dataset across several eukaryotic super-groups. In particular, the Alveolata displayed an unsuspected rDNA diversity in lineages such as Amoebophrya (MALV-II), Haematodinium (MALV-IV), Blastodinium (Dinophyceae), Parvilucifera (Perkinsea), Vampyrophrya (Ciliophora), and Cephaloidophoroidea (gregarine apicomplexans). MALVs sensu lato (>8,000 OTUs) are known parasitoids of dinoflagellates, rhizarians, ciliates, and metazoans (Bråte et al. 2012; L. Guillou et al. 2008; Massana et al. 2014; R. Siano et al. 2011). Parvilucifera infects dinoflagellates, and Cephaloidophoroidea (384 OTUs), Blastodinium and Vampyrophrya parasitize crustaceans (Skovgaard, Karpov, and Guillou 2012). Rhizaria also include significant parasites/parasitoids, with 160 OTUs in Ascetosporea (parasites of invertebrates including Haplosporidia, Paramyxea, and Paradinium) and 196 and 53 OTUs related to Cryothecomonas and Pseudopirsonia, respectively (parasitoids of diatoms)(Burki and Keeling 2014). Importantly, the excavate Diplonemida have a known parasitic lifestyle in other biomes (Elston and Sawyer 1987; von der Heyden et al. 2004; Schnepf 1994). Their huge diversity unveiled herein could represent another very significant reservoir of parasitic diversity undescribed in marine plankton. Thus, while hyper-diverse groups of relatively large eukaryotic plankton such as diatoms, metazoans, or rhizarians may escape predatory pressure thanks to their size and/or skeletons, they can be infected by a wide range of parasites which likely regulate their populations and have co-diversified with their host.
Figure W1. The Tara Oceans expedition (Sept. 2009 - March 2012) and the 47 sampling stations analyzed in the ‘Global Oceans Eukaryotic Plankton Diversity’ paper.At each station, eukaryotic plankton community was sampled at two depths (subsurface and Deep Chlorophyll Maximum (DCM), and fractionated into four main organismal size categories (0.8-5µm: "pico-nano"; 5-20µm: "nano"; 20-180µm: "micro"; 180-2000µm: "meso"-plankton). Except for a short incursion into the Southern Ocean at stations TARA_082 to TARA_085, our data do not concern plankton biodiversity in polar oceans.
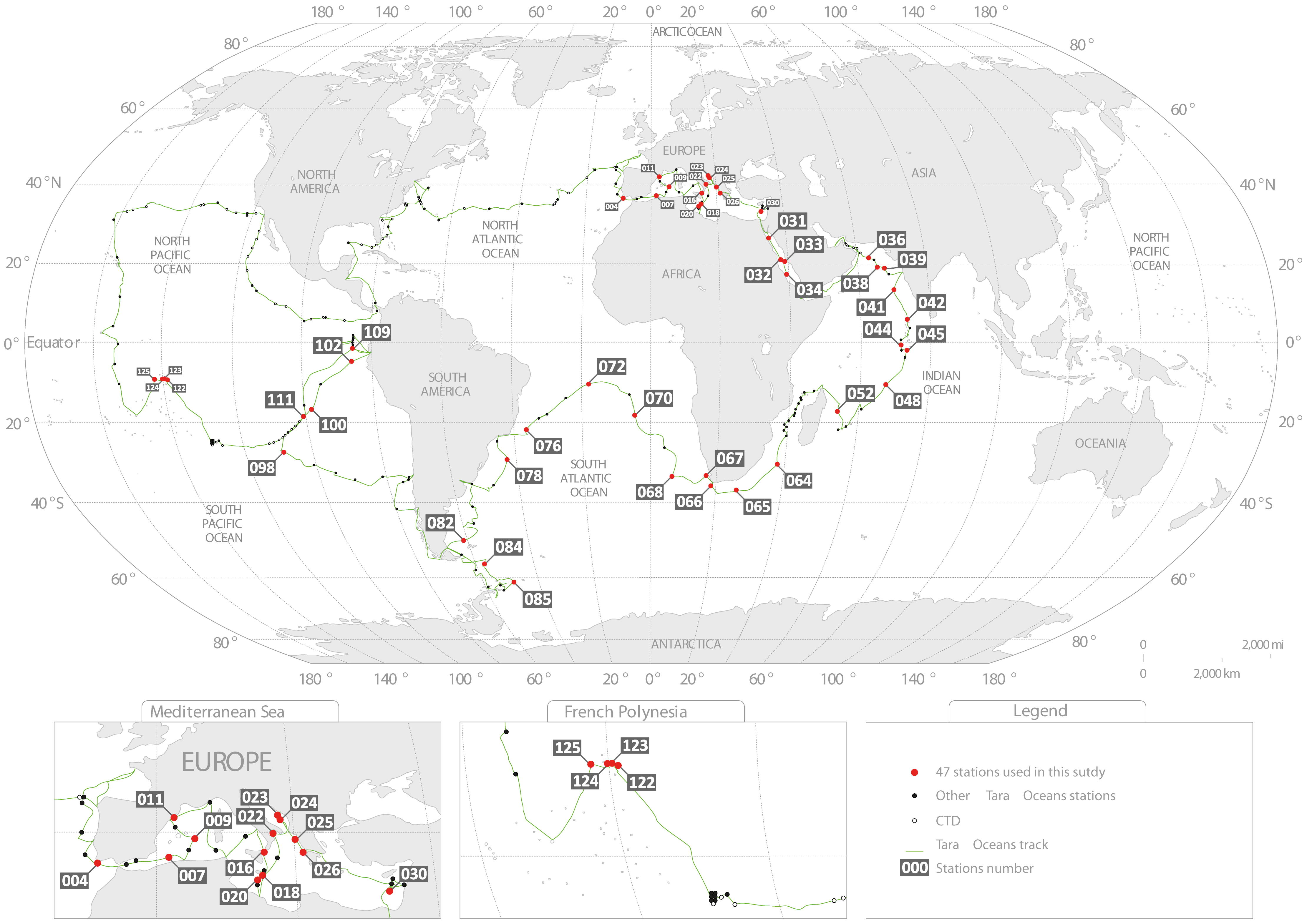
Figure W2. Bioinformatics pipeline and OTUs’ taxonomic purity. A: Raw V9 rDNA reads were first filtered based on sequence quality scoring and chimera removal analyses, and only reads present in at least 3 copies and 2 independent samples were considered for downstream analyses. Filtered reads were dereplicated and taxonomically assigned by homology (ggsearch global alignment) to an expert-curated database (the V9_PR2*, see databases W2 and W3). Metabarcodes -identical dereplicated reads- were finally clustered into OTUs (Operational Taxonomic Units) using the Swarm algorithm (Mahé et al. 2014) for subsequent α- and β-diversity analyses. B: Tara Oceans metabarcodes were clustered into biologically meaningful OTUs, using a 1 bp difference (local threshold) (Mahé et al. 2014). The 'swarming’ procedure has the advantage of avoiding arbitrary global clustering thresholds and input-order dependency induced by centroid selection, a typical bias of classical clustering methods. Although it has the potential to form large chains of barcodes, this is rarely the case and the large majority of Tara Oceans OTUs were indeed discrete entities with a single and consistent taxonomic assignment. Each barcode within each OTU received a taxonomic assignment. In order to compute OTU’s taxonomic purity, we first identified the dominant taxonomic assignment in each OTU, i.e., the one that recruits the greatest number of reads. That dominant assignment is then compared to the total number of reads in the OTU to compute a % value. The left panel shows the distribution of OTU purities (defined as the % of reads within an OTU assigned to the same taxon) as a function of the OTU masses (i.e., the total number of reads found in each OTU) for all OTUs containing ≥100 reads. OTUs are colored according to the identity of their most abundant barcode to reference sequences, from dark to light blue from low to high identity, respectively. On the right panel, the distribution of Tara Oceans OTU purities shows that the vast majority of OTUs (~88%) are ‘pure’. With their large radii, the largest OTUs are the most likely to present low purities, but remarkably only 8 out of 87 OTUs containing 106 or more reads have a purity <75%. As expected, an important proportion of low purity OTUs is made of OTUs with a low percentage of identity to reference sequences (colored in dark blue). Low identity taxonomic assignments tend to be less robust to small differences in nucleotidic sequences, which can artificially lower purity values.
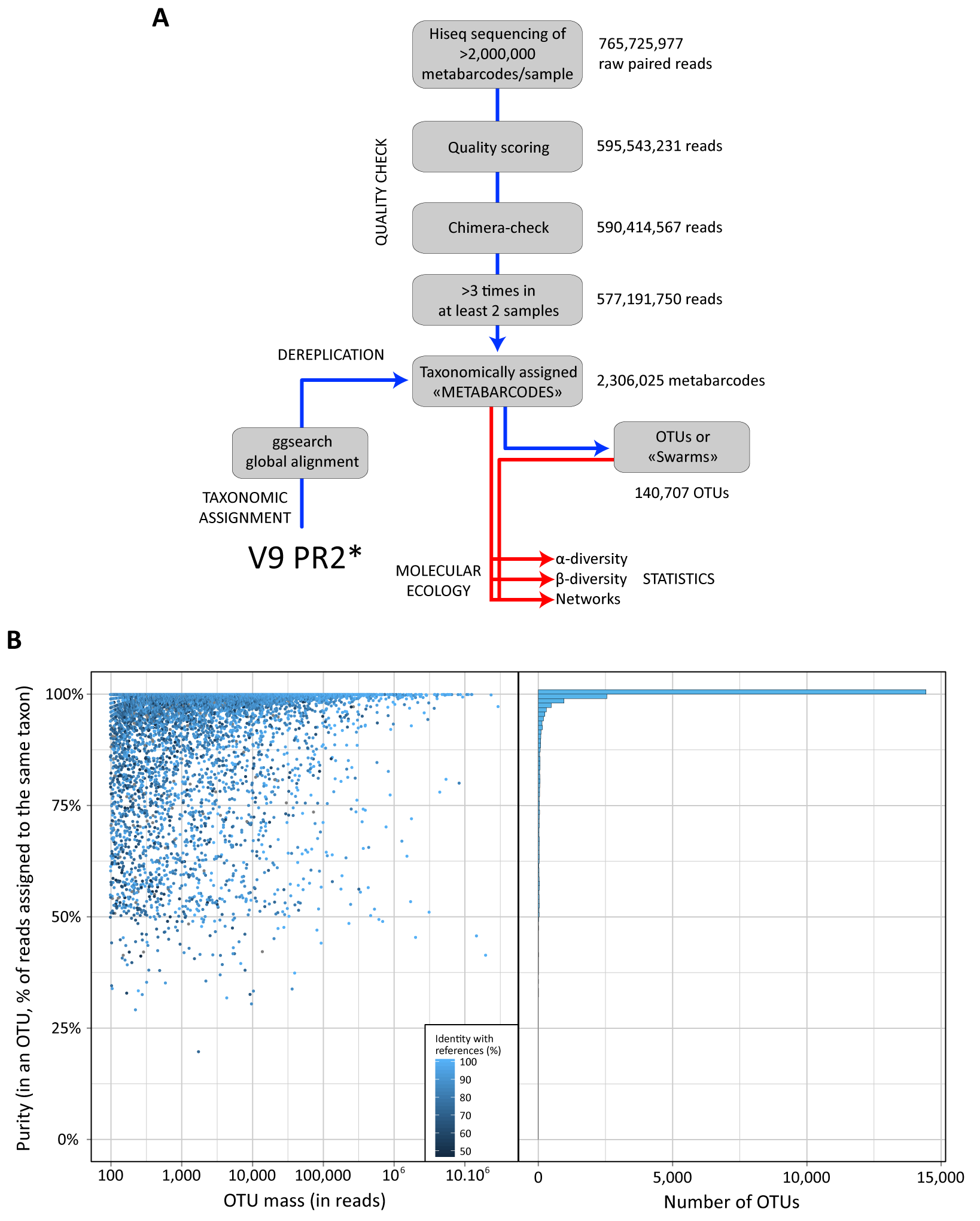
Figure W3. V9 rDNA as a crude proxy for relative biovolume in eukaryotes. A: Correlation between rDNA copy number and organism length/size across a wide taxonomic diversity of eukaryotes (including phototrophic and heterotrophic protists, as well as metazoan copepods) covering an organismal size spectrum equivalent to the one sampled in this study, from 0.8 µm to 2,000 µm. Data were gathered from published studies comparing cell length and rDNA copy numbers, including a wide range of protistan and metazoan species from various eukaryotic lineages such as dinoflagellates (Godhe et al. 2008; Zhu et al. 2005), ciliates (Gong et al. 2013), foraminifers (Weber and Pawlowski 2013), chlorophytes (Zhu et al. 2005), diatoms (Godhe et al. 2008; Zhu et al. 2005), or crustaceans (Wyngaard et al. 1995). Note that for metazoan taxa (copepods), rDNA copy numbers were multiplied by the approximate number of cells found in a copepod (~10,000). The equation of the linear correlation between cell length and rDNA copy number is shown. Ciliates were excluded from this specific analysis, because they are clearly outliers whose rDNA abundance counts are greatly affected by their unique dual genome architecture and differential gene amplification patterns, which can disassociate rDNA abundances from cell volume. Most of the ciliate data come from the study of Gong et al. (2013). Their results were exceedingly high compared to previous studies of ciliates, which is likely due to their methods measuring miniprepped DNA rather than DNA levels within individual cells. Nevertheless, DNA copy in general, and rDNA in specific, are known to vary substantially amongst ciliate taxa such that smaller species can sometimes have much higher rDNA copies than larger cells (Dunthorn et al. 2014). B: Comparison between light microscopy-based biomass, biovolume, count data, and V9 rDNA read number for different eukaryotic phytoplankton groups from Tara Oceans micro-plankton samples collected in surface waters from the Indian, Atlantic, and Southern Oceans (Stations TARA_052 to TARA_082, see Fig. W1). C: Comparison between light microscopy counts and V9 rDNA read numbers of different diatom genera (Tara Oceans stations and samples as in B.).
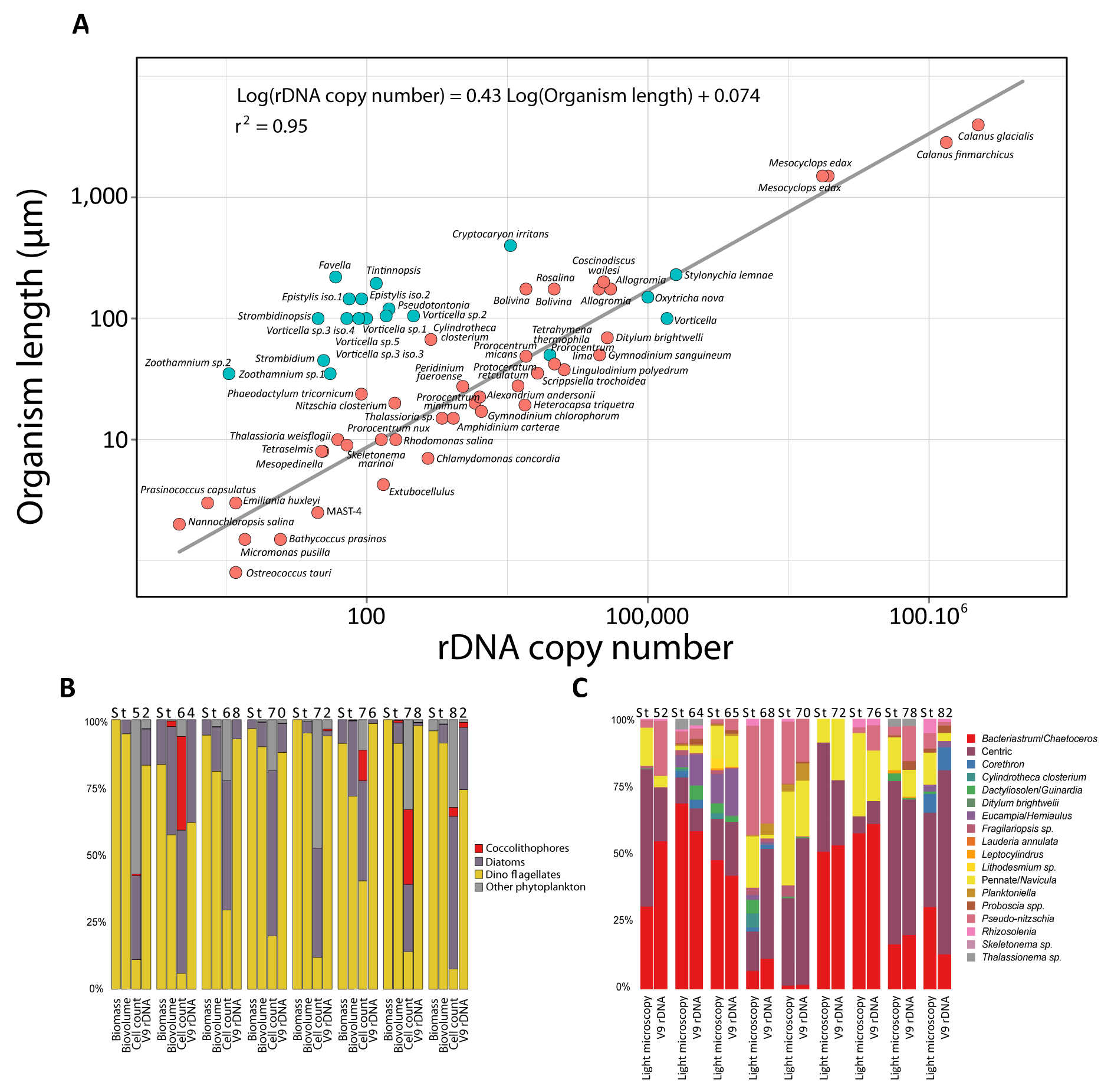
Figure W4. Saturation and richness of size-fractionated eukaryotic V9 rDNA metabarcodes and OTUs from the world photic oceans. A: Saturation curve for metabarcodes richness from the different eukaryotic plankton size-fractions; B: Overall (all samples) richness per organismal size-fraction, based on normalized size samples.
Figure W5. Similarity of Tara Oceans rDNA richness and abundance to total referenced eukaryotic rDNA diversity available in public databases. Abundance (Y-axis) and % identity to best reference barcode (X-axis) for all Tara Oceans V9 rDNA OTUs (left panel) and reads (right panel). Proportion of OTUs and reads per eukaryotic super-group is color-coded.
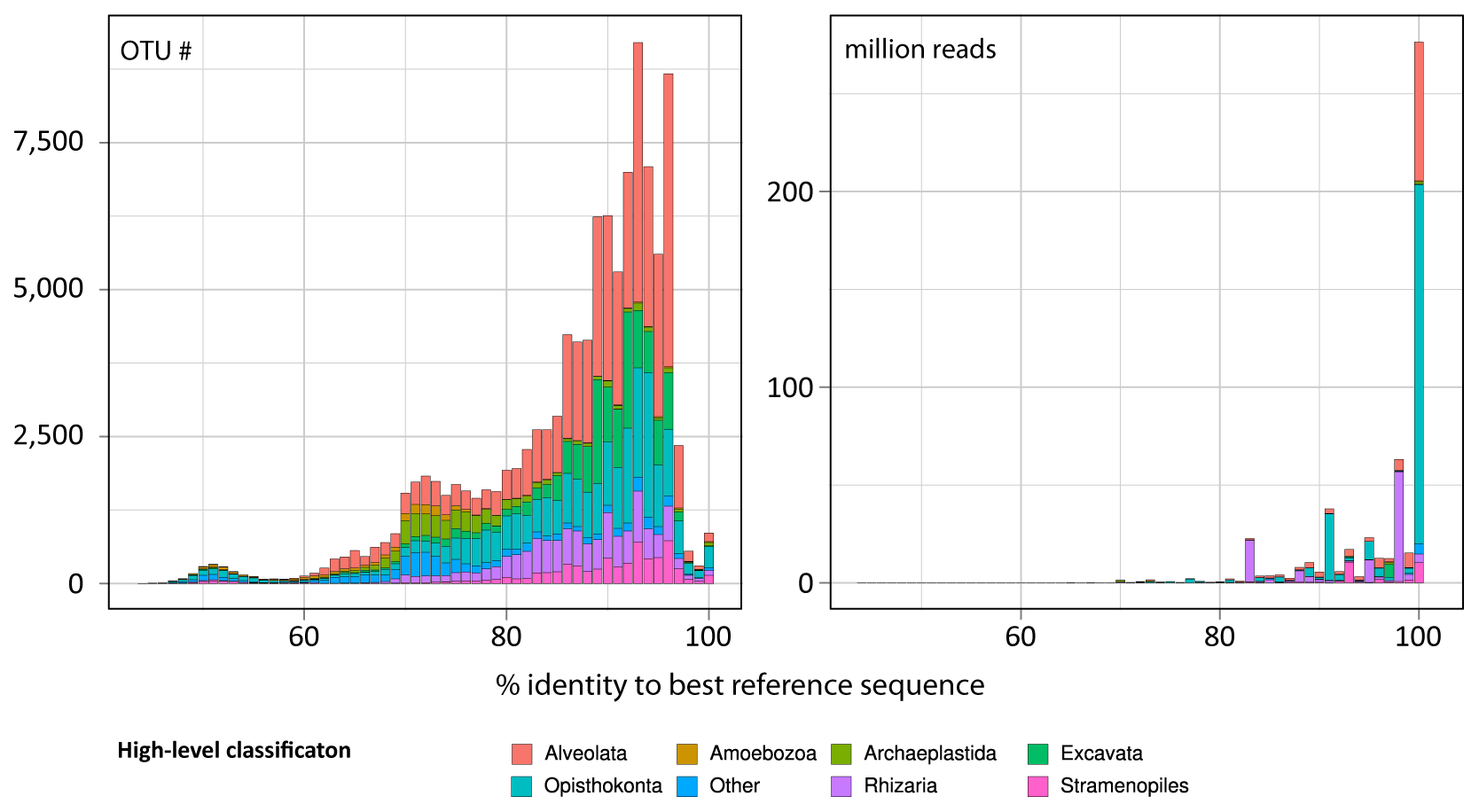
Figure W6. Phylogenetic novelty provided by Tara Oceans metabarcodes to prior knowledge of protistan rDNA sequences. A: Tree-length increase (%) after addition of the Tara Oceans V9 rDNA metabarcodes to reference trees, as a measure of the increase of phylogenetic information generated by the Tara Oceans dataset for each major eukaryotic lineage (see Detailed Material & Methods). The highest increase was found in diplonemids, a group of small-sized heterotrophic and occasionally parasitic organisms. The giant colonial radiolarian group Collodaria followed, with an increase of 3,560% in spite of the large size of its members. In third position came the exclusively parasitic group Perkinsea, illustrating the immense diversity of parasitoids unveiled in oceanic plankton. Groups that did not increase substantially their tree length include sediment-associated amoeboid, sessile or mycelial organisms, as well as certain highly specialized groups of parasites. B: Phylogenetic novelty per eukaryotic supergroup. The highest increase was found in Alveolata. Their unknown diversity concerns not only parasitoid groups (Perkinsea, MALV I-V), but also the supposedly well–characterized dinoflagellates. On the other hand, the super-phylum Amoebozoa, comprising almost exclusively organisms living on substratum, did not show a substantial increase in diversity.
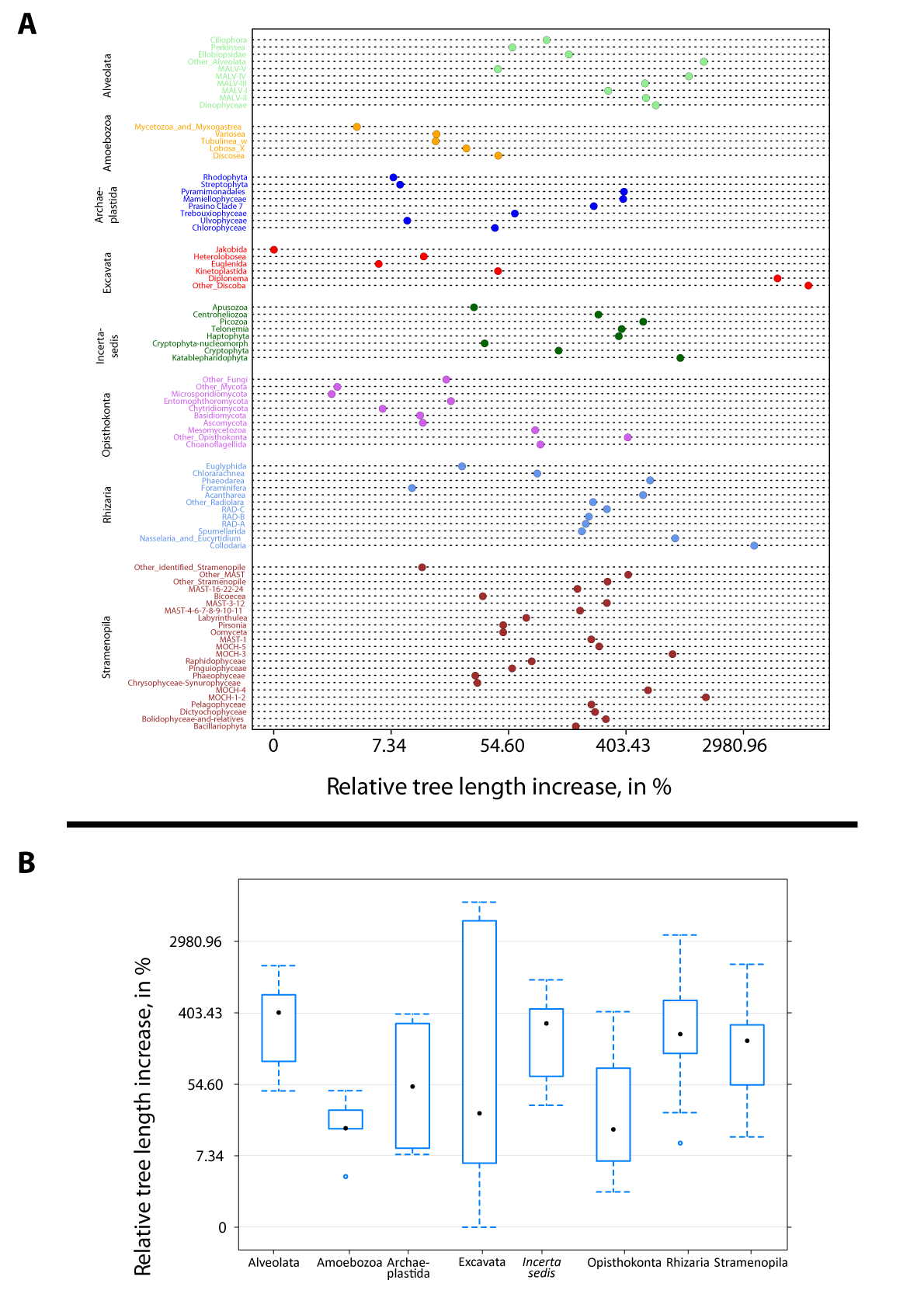
Figure W7. Example of the phylogenetic novelty brought by Tara Oceans metabarcodes in classical and conspicuous groups of planktonic protists. A: Diatoms (Bacillariophyta; Stramenopila), arguably the most studied group of microalgae. B: The heterotrophic Acantharia (Radiolaria, Rhizaria). Phylogenetic trees on the left with red terminal branches contain only reference V9 rDNA barcodes (database W2), while trees on the right contain both reference barcodes (red branches) and Tara Oceans metabarcodes (blue branches, one representative sequence for each OTU, see Detailed Material & Methods). The new Tara Oceans metabarcodes expanded considerably our vision of the diversity of these well known planktonic groups which form complex external mineral structures of amorphous silica (diatoms) and strontium sulfate (acantharians), used as morphological diagnoses for species identification. Marine planktonic diatoms are estimated to encompass ~164 genera (and ~1,800 species, (Sournia, Chrétiennot-Dinet, and Ricard 1991)), a large number of them having been barcoded over the last two decades. Contrary to the expectation that most of the diatom diversity has already been described, our data revealed not only a wealth of potential new species in known groups, but also novel, relatively deep branching clades. The uncultured Acantharia are amongst the most conspicuous and largest (from 50 µm up to 5 mm) marine planktonic protists. Since their first morphological observations in the 19th century, this group has remained understudied, with only 160 morphospecies described (Decelle, Suzuki, et al. 2012; Schewiakoff 1926). The Tara Oceans metabarcodes generated >1,000 acantharian OTUs, including many deep branching clades, which suggests the existence of unsuspected novel groups and morphotypes, with potentially naked or small-sized forms. Both reference- and reference + Tara Oceans sequences-based trees were generated for each major planktonic lineage (database W9).
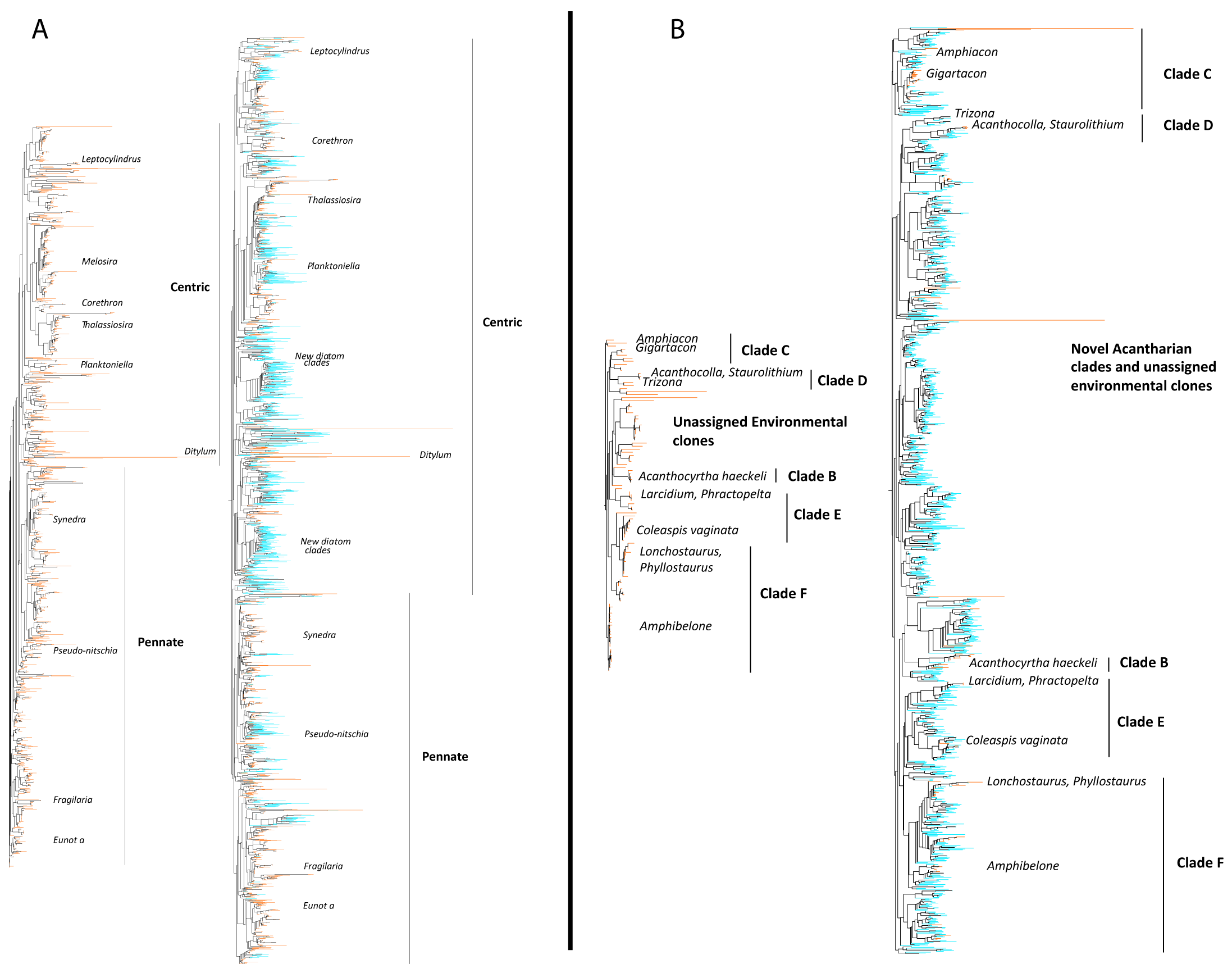
Figure W8. Broad functional categorization of the 11 hyper-diverse planktonic eukaryotic lineages. Pie-charts displaying the contribution of the 11 hyper-diverse eukaryotic lineages to broad ecological functions: parasitism ("Parasites"), phagotrophy ("Phago"), phototrophy ("Auto"), mixotrophy ("Mixo"), in terms of richness (number of OTUs, A) and abundance (B). Richness is based on total OTU number from all size-fractions, while abundance is based on read number from all piconano-plankton samples, which is the closest fraction to non-fractionated samples in terms of community composition (Fig. 6, main paper). Dotted line indicates ‘hypothetical’ function (in the case of diplonemids, see text W5).
Figure W9. rDNA-based abundance and diversity of main trophic modes across organismal size-fractions in photic-zone eukaryotic plankton. Box plots showing, across five organismal size fractions, the relative abundance (A) and diversity (B) of V9 rDNA metabarcodes assigned to phagotrophs (without chloroplasts), parasites, phototrophs (with permanent chloroplasts) and obligatory photosymbiotic hosts (hosting symbiotic microalgae). Calculations were based on all Tara Oceans samples from surface waters with, respectively, 17, 40, 21, 41, and 42 samples for the [0,8 µm-inf], [0,8-5 µm], [5-20 µm], [20-180 µm], and [180-2000 µm] organismal size fractions. The functional categories are exclusive, meaning that a given OTU can only belong to one of them. The last category "NA" contains the V9 rDNA metabarcodes which were functionally undefined according to our criteria.
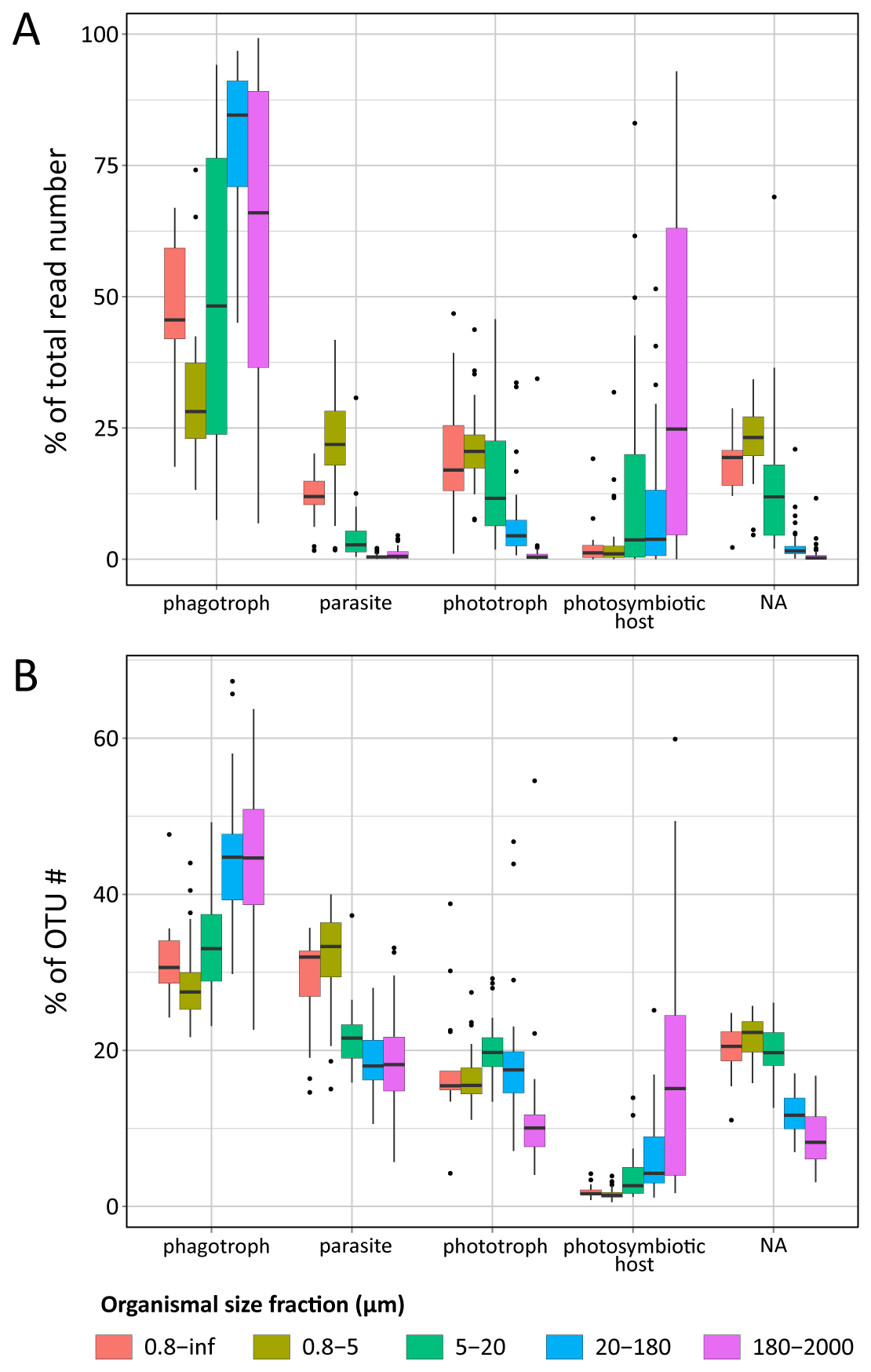
Figure W10. Most abundant eukaryotic groups (A, B), known eukaryotic symbionts, sensu lato (B, C), and eukaryotic phytoplankton (D, E), based on total rDNA reads and OTUs across organismal size fractions, depths, and geography. Bar charts display the relative abundance and richness (%) of V9 rDNA metabarcodes across Tara Oceans stations. Data are shown separately for the different organismal size fractions and sampling depths (surface or DCM waters), and the 47 Tara Oceans stations are separated into 7 oceanographic basins: NAtlO: North Atlantic Ocean; MedS: Mediterranean Sea; RedS: Red Sea; IndO: Indian Ocean; SAtlO: South Atlantic Ocean; SouO: Southern Ocean; PacO: Pacific Ocean. A, B: Overall, taxonomic and functional groups were more stable across space and time in the piconano- plankton, in terms of both metabarcode richness and abundance, suggesting that larger-sized planktonic taxa are less homogeneously distributed across the world oceans and can increase their population substantially when conditions allow. Furthermore, taxo-functional richness was less variable across stations than its abundance, implying that the pool of taxa within a given function is relatively constant across time and space, while some peak locally when conditions are favorable. C, D: Relative contribution of the most important groups of known parasites and mutualist microalgae amongst the entire community of symbionts, sensu lato. Note the clear shift in taxonomic composition between the piconano- and the meso-plankton, where for instance the known photosymbionts of pelagic rhizarians (the dinoflagellates Brandtodinium (Probert et al. 2014) and Pelagodinium (Shaked and de Vargas 2006; Raffaele Siano et al. 2010) for the collodarians and foraminifers, respectively, and the haptophyte Phaeocystis for the Acantharia (Decelle, Probert, et al. 2012)) are particularly apparent. E, F: Relative contribution (%) of the most abundant phytoplankton taxa (V9 rDNA metabarcodes) to total eukaryotic phytoplankton across Tara Oceans stations.



Figure W11. Potential technical impact on the Tara Oceans rDNA metabarcoding dataset: plankton size fractionation and whole genome amplification. B: Phylogenetic distribution of Tara Oceans V9 rDNA OTUs restricted to a single organismal size fraction. (a) and (b) as in Fig. 3 (main paper); (c): % of OTUs restricted to a single organismal size fraction for each major eukaryotic lineage; (d): % of size-restricted OTUs per plankton organismal size fraction (light blue = piconano-; green = nano-; yellow = micro-; red = meso-plankton). Overall, ~36% of OTUs were restricted to a single organismal size fraction, which is surprisingly high considering (i) the ontogenic and life-cycle stages of various sizes that characterize most eukaryotic taxa, and (ii) the potential mixing between size fractions occurring during the filtration/sieving process, due to either detritus of larger cells/organisms contaminating smaller size-fractions, or aggregates of small cells/organisms contaminating larger organismal size-fractions. In particular, in major groups known to be relatively large (metazoa, collodaria, phaeodarea, bacillariophyta), the majority of size-specific OTUs were part of the meso- or micro-plankton, indicating minor contamination from the larger to the smaller size-fractions. A: Bias in whole-genome amplified samples. Community composition dissimilarity (Bray-Curtis distances visualized using Non-linear Multi-Dimensional Scaling) amongst all samples (symbols), including those subject to whole genome amplification (wga, black-dot inside colored symbol) when extracted total DNA was not sufficient for metagenomic sequencing. Both size fractions (p-value = 10-3, r2= 0.55) and whole genome amplification (p-value = 10-3, r2 = 0.077) had significant impact on community structuring. Removing wga samples increased the significance of the effects of organismal size fraction on community structuring (p-value = 10-3, r2 = 0.73, see also Fig.6A main paper). WGA samples were thus removed from all subsequent β-diversity analyses. Shape and colors of symbols code for sampling depths and organismal size fractions, respectively.
Figure W12. Analysis of community differentiation vs. geographic distance in different plankton organismal size fractions. Scatter-plots of community differentiation (Bray-Curtis dissimilarities) and geographic distances (km) between communities are shown for each organismal size fraction. Geographic distances were obtained as great circle distances avoiding lands between sampling locations, computed using a least-cost distance strategy as implemented in the 'gdistance' R package (http://CRAN.R-project.org/package=gdistance). Each main panel represents a partial view of the scatter-plot for which there is a significant positive correlation (represented by the blue line). The inner left panels show a view of the scatter-plot over the entire range of geographic distances, with a highlighted area corresponding to the range displayed in the main panel. The inner right panels display Mantel correlogram between Bray-Curtis and geographic distance matrices. Main panel scatter-plots show a significant positive correlation at distances below ~6,000 km in all size fractions, a correlation that vanishes for higher distances (inner panels), as indicated by the Mantel R statistic (computed using the 'vegan' R package). For distances within 6,000 KM, the positive correlation increases with increasing organismal size fraction (p-value = 10-3, R=0.36, 0.49, 0.50, 0.51 from piconano- to meso-plankton respectively).
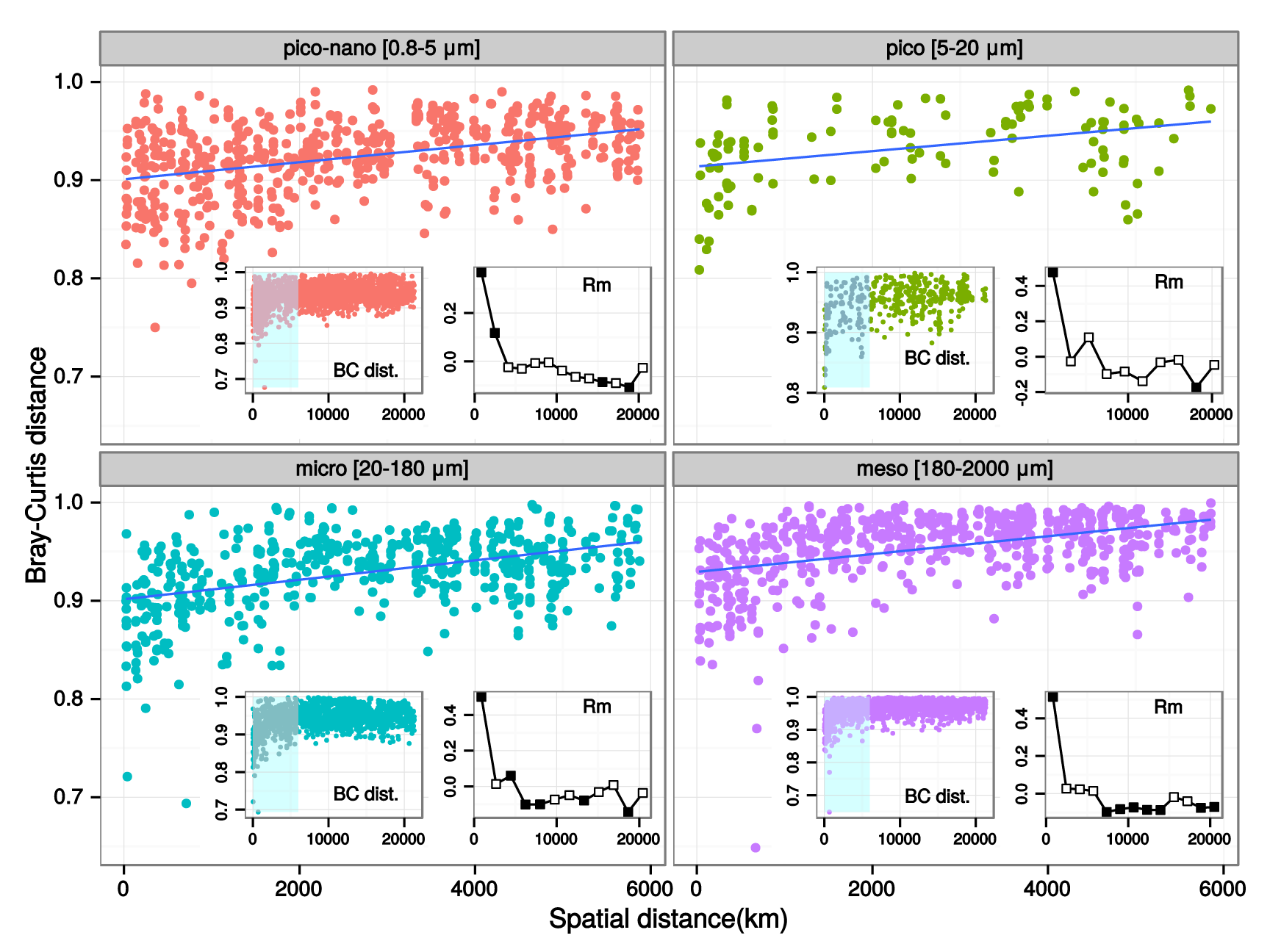
Figure W13. Abundant and rare eukaryotic plankton OTUs in all samples. Proportions of abundant (red dots) and rare (blue dots) OTUs across all analyzed samples and 4 eukaryotic plankton organismal size fractions. Subsampling was performed to normalize the number of reads per sample. Locally abundant and rare OTUs were then defined as those containing >1% and <0.01% of the reads in a given sample, respectively.
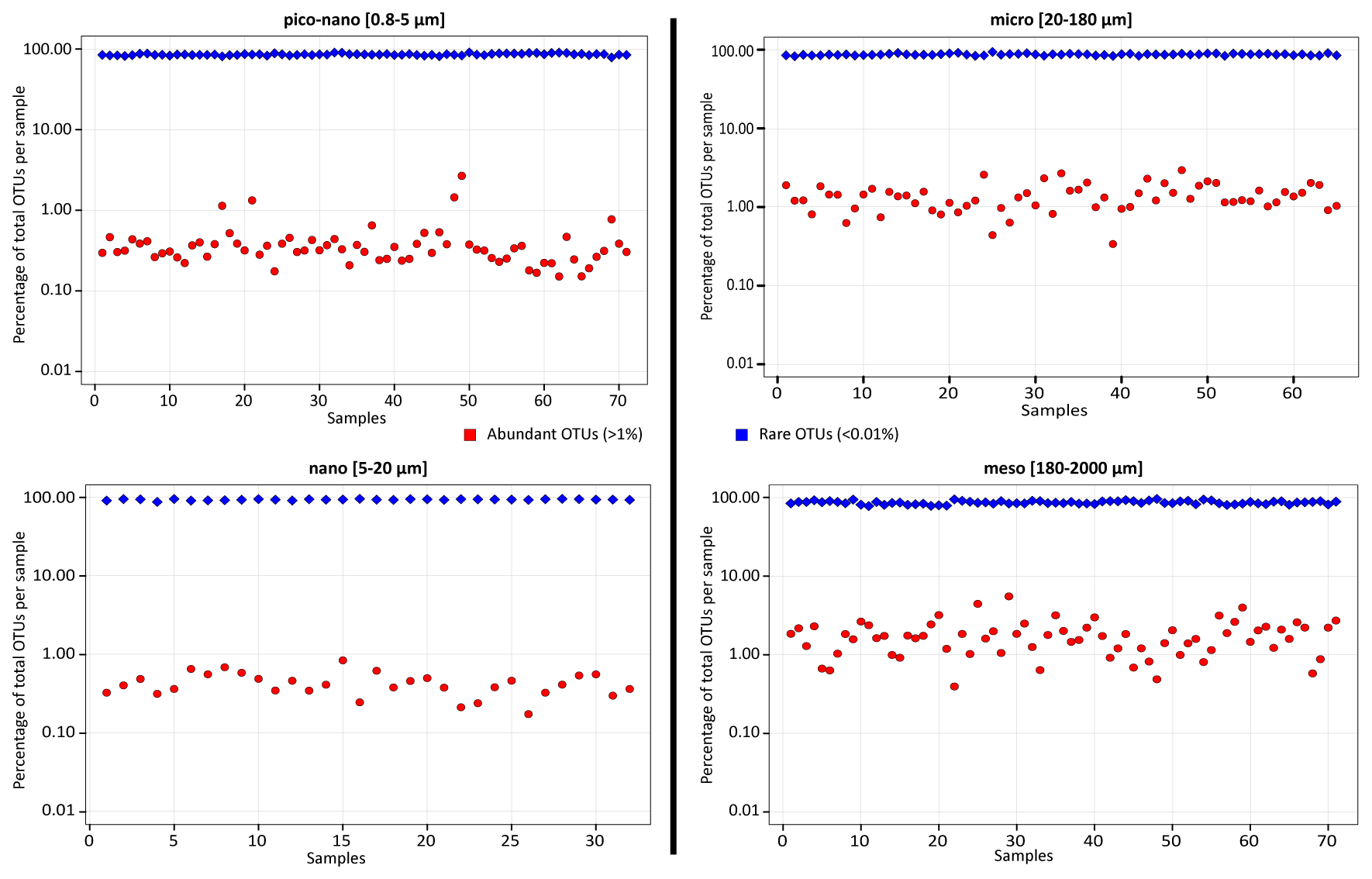
Figure W14. Metabarcoding inference of trophic/symbiotic modes of photic-zone eukaryotic plankton. Ecological richness (OTU number, left panel) and abundance (read number, right panel) of Tara Oceans rDNA metabarcodes assigned with at least 99% of identity to reference sequence of a given trophic/symbiotic mode. This analysis shows that the distribution patterns of trophic/symbiotic modes across geography and organismal size fractions are very similar, whether one use a 80% (Fig.5A in de Vargas al. submitted) or 99% similarity cutoff, demonstrating their robustness across evolutionary times. The main difference between both approaches is the decrease of rDNA OTUs richness affiliated to photosymbiotic protistan hosts in the larger size-fraction (mesoplankton), which is due to the fact that the large majority of these metabarcodes (Fig.5A in de Vargas al. submitted) are affiliated to the Collodaria, a complex and highly-diverse group of obligatory photosymbiotic rhizarians (>5,600 OTUs revealed in this study, see Fig.3 in de Vargas al. submitted) which contains only 81 reference sequences.

Database W1. The 334 size fractionated eukaryotic plankton community samples analysed herein, with a suite of associated metadata (Excel format). Note that if most samples represented the piconano- (0.8-5 µm, 73 samples), nano- (5-20 µm, 74 samples), micro- (20-180 µm, 70 samples), and meso- (180-2000 µm, 76 samples) planktonic size fractions, some represented different organismal size-fractions: 0.2-3 µm (1 sample), 0.8-20 µm (6 samples), 0.8 µm – infinity (33 samples), and 3-20 µm (1 sample). The table contains the following fields: a unique sample sequence identifier; the sampling station identifier; a Pangaea (http://www.pangaea.de) accession number identifying the genetic sample from which the sequences were obtained; an INDSC accession number allowing to retrieve raw sequence data for the major nucleotide databases (short read archives at EBI, NCBI or DDBJ); the depth of sampling (Subsurface – SUR or Deep Chlorophyll Maximum – DCM); the targeted size range; the sequences template (either DNA or WGA/DNA if DNA extracted from the filters was Whole Genome Amplified); the latitude of the sampling event (decimal degrees); the longitude of the sampling event (decimal degrees); the time and date of the sampling event; the device used to collect the sample; the logsheet event corresponding to the sampling event ; the volume of water sampled (liters). Then follows information on the cleaning bioinformatics pipeline (Fig.W2): the number of merged pairs present in the raw sequence file; the number of those sequences matching both primers; the number of sequences after quality-check filtering; the number of sequences after chimera removal; and finally the number of sequences after selecting only barcodes present in at least three copies in total and in at least two samples. Finally, are given for each sequence sample: the number of distinct sequences (metabarcodes); the number of OTUs; the average number of barcode per OTU; the Shannon diversity index based on barcodes for each sample (database W4); and the Shannon diversity index based on each OTU (database W5). Due to its general use, this dataset is archived at Pangaea under doi http://doi.pangaea.de/10.1594/PANGAEA.843017
Download (Size: 211 KB)Database W2. V9_PR2. The V9 rDNA Protistan Ribosomal Reference database, in fasta format, contains 77,449 reference V9 rDNA barcodes representing 13,432 genera and 24,435 species from all known major lineages of the tree of eukaryotic life. The header line of each reference V9 rDNA barcode (with a > sign) contains a unique identifier derived from GenBank accession number, followed by the taxonomic path associated to the reference barcode.
Download (Size: 18 MB)Database W3. V9_PR2*. The subset of V9_PR2 reference barcodes that recruited Tara Oceans metabarcodes, named at the genus level, and annotated with basic eco-bio/logical functions (Tab separated text format). Each reference barcode is associated with a sequence identifier (same as in database W2) and three categories of functional annotations: (1) Chloroplast: yes, presence of permanent chloroplast; no, absence of permanent chloroplast ; NA, undetermined. (2) Symbiont (small partner): parasite, the species is a parasite; commensal, the species is a commensal; mutualist, the species is a mutualist symbiont, most often a microalgal taxon involved in photosymbiosis; no the species is not involved in a symbiosis as small partner; NA, undetermined. (3) Symbiont (host): photo, the host species relies on a mutualistic microalgal photosymbiont to survive (obligatory photosymbiosis); photo_falc, same as photo, but facultative relationship; photo_klep, the host species maintains chloroplasts from microalgal prey(s) to survive; photo_klep_falc, same as photo_klep, but facultative; Nfix, the host species must interact with a mutualistic symbiont providing N2 fixation to survive; Nfix_falc, same as Nfix, but facultative; no, the species is not involved in any mutualistic symbioses; NA, undetermined. For example, the collodarian/Brandtodinium symbiosis (Probert et al. 2014) is annotated: Chloroplast, "no"; Symbiont (small), "no"; Symbiont (host), "photo", for the collodarian host; and: Chloroplast, "yes"; Symbiont (small), "mutualist"; Symbiont (host), "no", for the dinoflagellate microalgal endosymbiont.
Download (Size: 1.3 MB)Database W4. Total V9 rDNA information organized at the metabarcode level. Tab separated text file in a zip archive listing for each Tara Oceans V9 rDNA metabarcode, and including the following fields: md5sum = unique identifier; lineage = taxonomic path associated to the metabarcode; pid = % identity to the closest reference barcode from V9_PR2; sequence = nucleotide sequence of the metabarcode; refs = identity of the best hit reference sequence(s); TARA_xxx = number of occurrences of this barcode in each of the 334 samples; totab = total abundance of the barcode ; cid = identifier of the OTU to which the barcode belongs; taxogroup = high-taxonomic level assignation of this barcode (see Material and Methods); chloroplast = "yes", "no" or "NA" (see legend database W3) ; symbiont.small = "parasite", "commensal", "mutualist", "no" or "NA" (see legend database W3); symbiont.host = "photo", "photo_falc", "photo_klep", "Nfix", no or NA (see legend database W3) ; benef = "Nfix", "no" or "NA" (see legend database W3); trophism = Metazoa , heterotroph , NA , photosymbiosis , phototroph according to the previous fields. Due to its general use, this dataset is archived at Pangaea under doi http://doi.pangaea.de/10.1594/PANGAEA.843018
Download (Size: 265 MB)
Database W5. Total V9 rDNA information organized at the OTU level. Tab separated text file compressed in a zip archive listing for each Tara Oceans V9 rDNA OTU the following fields: md5sum = identifier of the representative (most abundant) sequence of the swarm; cid = identifier of the OTU; totab = total abundance of barcodes in this OTU; TARA_xxx = number of occurrences of barcodes in this OTU in each of the 334 samples; rtotab = total abundance of the representative barcode; pid = percentage identity of the representative barcode to the closest reference sequence from V9_PR2; lineage = taxonomic path assigned to the representative barcode ; refs = best hit reference sequence(s) with respect to the representative barcode ; taxogroup = high-taxonomic level assignation of the representative barcode; chloroplast = "yes", "no" or "NA" (see legend database W3) ; symbiont.small = "parasite", "commensal", "mutualist", "no" or "NA" (see legend database W3); chloroplast = "yes", "no" or "NA" (see legend database S3); symbiont.host = "photo", "photo_falc", "photo_klep", "Nfix", no or NA (see legend database W3) ; benef = Nfix, NA or no (see legend database W3) ; trophism = Metazoa , heterotroph , NA , photosymbiosis , phototroph according to the previous fields. Due to its general use, this dataset is archived at Pangaea under doi http://doi.pangaea.de/10.1594/PANGAEA.843022
Download (Size: 17 MB)
Database W6. Table listing the 97 deep-branching eukaryotic morpho-lineages we took into account to assign Tara Oceans metabarcodes, and comprising all described eukaryotic diversity on Earth. These morpho-lineages correspond to higher-level categories in the V9_PR2 reference database and cover the entire database. The table provides the following information about each of them: (1) whether it contains at least some described marine planktonic species; (2) whether it has been found previously in marine plankton by environmental sequencing; (3) whether it is classically regarded as a significant group in marine plankton studies♯; (4) the known main trophic mode(s) of its members; (5) the known preferential habitat(s) of its members; (6) the number of reference sequences in V9_PR2; (7) the number of V9 rDNA reads assigned to that morpho-lineage in the Tara-Oceans data; (8) the number of OTUs assigned to that morpho-lineage in the Tara-Oceans data; and (9) additional remarks about each morpho-lineage, detailing in some cases which significant organisms within it are particularly abundant, or on the contrary not found at all in the Tara-Oceans data, and for those that correspond to taxonomically artificial groups, which organisms were included in it. The twelve morpho-lineages virtually absent from the Tara-Oceans data are shaded in grey. 85 were present in the world photic-ocean plankton and are represented in Fig. 3. The eleven 'hyper-diverse' lineages with more than 1,000 OTUs in the Tara Oceans data are highlighted in yellow. ♯In order to be classified as ‘classically ecologically significant’, a lineage should: (i) have a ‘yes’ in at least (1) or (2); (ii) be independently considered as classically ecologically significant by all protist experts co authoring the paper; (iii) for debatable case, be reported in a significant number of publications or included in models of plankton ecology.
Download (Size: 60 KB)
Database W7. Heatmap of the distribution of the 381 cosmopolitan OTUs across eukaryotic lineages, sampling depths, organismal size fractions, and Tara Oceans stations. OTUs are grouped by taxonomic lineage (see Fig. 3), and sorted by decreasing size (i.e., number of reads). Each OTU is represented by its rank number in the OTU table (database W5 and W8). The distribution of each OTU in the two different depths (surface and DCM), in the four size fractions (pico, nano, micro, meso) and in the 47 sampling stations is color-coded: the darker the tile, the more the OTU reads are concentrated in that station, depth or size fraction. The global distribution (sum of all distributions) added at the bottom of the figure indicates that the cosmopolitan OTUs are evenly distributed in the different depths, size fractions and stations. Because the largest cosmopolitan OTUs are evenly distributed (see the Metazoa and Dinophyceae groups), their weight dominates and smoothens the global distribution. Very contrasted patterns emerge for the different taxonomic groups. For instance, OTUs assigned to Haptophyta are clearly concentrated in the surface waters and in the smallest size fraction (piconano), and are, with a few exceptions, evenly distributed in the 47 sampling stations. OTUs assigned to Collodaria are more abundant in the large size fraction (meso) and are less evenly distributed, with spikes of abundance in certain sampling stations
Download (Size: 244 KB)
Database W8. Abundance, distribution and taxonomic assignments of the 381 cosmopolitan OTUs. Tab separated text file (zip archive) listing for each of the 381 cosmopolitan OTUs, including the following fields: md5sum = identifier of the representative (most abundant) sequence of the swarm; cid = identifier of the OTU; totab = total abundance of barcodes; 4 to 125 = number of occurrences of barcodes in each of the 47 stations; dataset% = share of the total dataset contained in that OTU; sum_X = number of occurrences of barcodes at each depth or size fraction; OTU_purity = percentage of the OTU barcodes sharing exactly the same taxonomic assignment; pid = percentage identity of the representative barcode to the closest reference sequence from V9_PR2; taxogroup = high-taxonomic level assignation of the representative barcode; lineage = taxonomic path assigned to the representative barcode.
Download (Size: 161 KB)
Database W9. Phylogenetic trees of reference and experimental sequences for the 85 major eukaryotic lineages. Phylogenetic trees (phyloxml files in a zip archive) including reference and experimental (Tara Oceans) sequences for each of the 85 major eukaryotic lineages (Fig. 3). See Material and Methods for methodological details. Visualisation of the trees with the Archaeopteryx software (https://sites.google.com/site/cmzmasek/home/software/archaeopteryx) allow color-coding of branches according to their origin (reference or Tara Oceans data) and weighting/coloring according to their abundance (# of reads for Tara Oceans sequences). Leafs corresponding to experimental sequences are named according to the following scheme: an unique identifier corresponding to entries in database W4 and W5 and identifying the OTU, the total abundance of the OTU, the percentage identity of the representative sequence of the OTU with respect to the best reference sequence and finally the taxonomic lineage assigned to it.
Download (Size: 3.6 MB )
Silvia G. Acinas1, Peer Bork2, Emmanuel Boss3, Chris Bowler4, Colomban de Vargas5,6, Michael Follows7, Gabriel Gorsky8,9, Nigel Grimsley10,11, Pascal Hingamp12, Daniele Iudicone13, Olivier Jaillon14,15,16, Stefanie Kandels-Lewis2, Lee Karp-Boss3, Eric Karsenti17,18, Uros Krzic19, Fabrice Not5,6, Hiroyuki Ogata20, Stephane Pesant21,22, Jeroen Raes23,24,25, Emmanuel Reynaud26, Christian Sardet8, Mike Sieracki27, Sabrina Speich28,29, Lars Stemmann8, Matthew B. Sullivan30, Shinichi Sunagawa2, Didier Velayoudon31, Jean Weissenbach14,15,16, Patrick Wincker14,15,16
1Department of Marine Biology and Oceanography, Institute of Marine Science (ICM)-CSIC, Barcelona, Spain. 2Structural and Computational Biology, European Molecular Biology Laboratory, Heidelberg, Germany. 3School of Marine Sciences, University of Maine, Orono, USA. 4Environmental and Evolutionary Genomics Section, Institut de Biologie de l’Ecole Normale Supérieure, Centre National de la Recherche Scientifique, Unité Mixte de Recherche 8197, Institut National de la Santé et de la Recherche Médicale U1024, Ecole Normale Supérieure, Paris, France. 5CNRS, UMR 7144, Station Biologique de Roscoff, Roscoff, France. 6Sorbonne Universités, UPMC Univ Paris 06, UMR 7144, Station Biologique de Roscoff, Roscoff, France. 7Dept of Earth, Atmospheric and Planetary Sciences, Massachusetts Institute of Technology, Cambridge, USA. 8CNRS, UMR 7093, LOV, Observatoire océanologique, Villefranche/mer, France. 9Sorbonne Universités, UPMC Univ Paris 06, UMR 7093, LOV, Observatoire océanologique, Villefranche/mer, France. 10CNRS UMR 7232, BIOM, Banyuls-sur-Mer, France. 11Sorbonne Universités, OOB, UPMC Paris 06, Banyuls-sur-Mer, France. 12Aix Marseille Université, CNRS, IGS UMR 7256, Marseille, France. 13Laboratory of Ecology and Evolution of Plankton, Stazione Zoologica Anton Dohrn, Naples, Italy. 14CEA, Genoscope, Evry France. 15CNRS, UMR 8030, Evry, France. 16Université d'Evry, UMR 8030, Evry, France. 17Environmental and Evolutionary Genomics Section, Institut de Biologie de l’Ecole Normale Supérieure, CNRS, UMR 8197, Institut National de la Santé et de la Recherche Médicale U1024, Ecole Normale Supérieure, Paris, France. 18Directors’ Research, European Molecular Biology Laboratory, Heidelberg, Germany. 19Cell Biology and Biophysics, European Molecular Biology Laboratory, Heidelberg, Germany. 20Institute for Chemical Research, Kyoto University, Kyoto, Japan. 21PANGAEA, Data Publisher for Earth and Environmental Science, University of Bremen, Bremen, Germany. 22MARUM, Center for Marine Environmental Sciences, University of Bremen, Bremen, Germany. 23Department of Microbiology and Immunology, Rega Institute KU Leuven, Leuven, Belgium. 24VIB Center for the Biology of Disease, VIB, Leuven, Belgium. 25Laboratory of Microbiology, Vrije Universiteit Brussel, Brussels, Belgium. 26School of Biology and Environmental Science, University College Dublin, Dublin, Ireland. 27Bigelow Laboratory for Ocean Sciences, East Boothbay, USA. 28Department of Geosciences, Laboratoire de Météorologie Dynamique (LMD), Ecole Normale Supérieure, Paris, France. 29Laboratoire de Physique des Océan, UBO-IUEM, Plouzané, France. 30Department of Ecology and Evolutionary Biology, University of Arizona, Tucson, USA. 31DVIP Consulting, Sèvres, France.Ameer Abdulla, Chantal Abergel, Denis Allemand, Aldine Amiel, Leif Anderson, David Antoine, Detlev Arendt, Roberto Arrigoni , Defne Arslan, Francois Artiguenave, Stephane Audic, Jean-Marc Aury, Marcel Babin, Celine Bachelier, Xavier Bailly, Andrew Baker, Cecilia Balestra, Benedetto Barone, Daniela Basso, Daniel Bayley, Gregory Beaugrand, Laurent Beguery, Elia Benito-Guttierez, Francesca Benzoni, Eric Beraud, Lionel Bigot, Lucie Bittner, Martine Boccara, Roxane Boonstra, Peer Bork, Emmanuel Boss, Christophe Boutte, Chris Bowler, Annick Bricaud, Jennifer Brum, Jeremie Capoulade, Luigi Caputi, Annalisa Caragnano, Margaux Carmichael, Raffaela Casotti, Ivona Cetinic, Samuel Chaffron, Aurélie Chambouvet, Patrick Chang, Ali Chase, Claudia Chica, Hervé Claustre, Jean-Michel Claverie, Camille Clerissi, Sebastien Colin, Montse Coll-Lladó, Steeve Comeau, Christian Conrad, Laurent Coppola, Miguel Francisco Cornejo, Marcella Cornejo , Daniel Cossa, Maryam Cousin, Corinne Cruaud, Corrine Cuck, Marcela D’Ottone, Corinne Da Silva, Denis Dausse, Denis de la Broise, Silvia De Monte, Colomban de Vargas, Johan Decelle, Alan Deidun, Javier del Campo, Evelyne Derelle, Yves Desdevises, Elodie Desgranges, Valerie Desplanches, Floriane Despres, Nicolas Desreumaux, Rosanna di Mauro, Celine Dimier, John Dolan, Fabrizio D'Ortenzio, Francesco d'Ovidio, Anne Doye, Melissa Duhaime, Emilie Duperche, Xavier Durrieu de Madron, Stephanie Dutkiewicz, Karoline Faust, Janine Felden, Beatriz Fernández, Isabel Ferrera, Regis Ferriere, Christine Ferrier-Pagès, Mick Follows, Rainer Friedrich, Françoise Gaill, Alexandre Ganachaud, Laurence Garczarek, Josep M Gasol, Stéphane Gasparini, Jean-Pierre Gattuso, Gabriella Gilkes, Jennifer Gillette, Silvia G. Acinas, Gabriel Gorsky, Brett Grant, Nigel Grimsley, Jean-Michel Grisoni, Michel Groc, Lionel Guidi, Cedric Guigand, Luis Gutierrez-Herredia, Roland Hellig, Pascal Hingamp, Danwei Huang, Julio Ignacio-Espinoza, Daniele Iudicone, Olivier Jaillon, Jean-Louis Jamet, Stefanie Kandels-Lewis, Lee Karp-Boss, Eric Karsenti, Michael Katinka, Yuko Kitano, Zbigniew Kolber, Philippe Koubbi, Uros Krzic, Hironobu Kukami , Karine Labadie, Pamela Labbe-Ibanez, Tomas Larsson, Alban Lazar, Herve Le Goff, Corinne Le Quere, Brian Leander, Philippe Lebaron, Noan LeBescot, Thomas Lefort, Louis Legendre, Cristophe Lejeusne, Cyrille Lepoivre, Magali Lescot, Mangan Lewis, Edouard Leymarie, Gipsi Lima-Mendez, Ramiro Logares, Frédéric Mahé, Cornelia Maier, Shruti Malviya, Catarina Marcolin, Claudie Marec, Sophie Marinesque, Ramon Massana, Lydiane Mattio, Maria Grazia Mazzochi, Raphaël Morard, Hervé Moreau, Pascal Morin, Simon Morisset, David Mountain, Paul Muir, Harry Nelson, Sophie Nicaud, Paul Nival, Benjamin Noel, Fabrice Not, Grigor Obolensky, David Obura, Hiroyuki Ogata, Philippe Pages, Claude Payri, Javier Paz Yepes, Carlos Pedros-Alio, Eric Pelletier, Rainer Pepperkok, Fabien Perault, Yvan Perez, Stephane Pesant, Marc Picheral, Michel Pichon, Gwenaël Piganeau, Ruby Pillay, Olivier Poirot, Julie Poulain, Nicole Poulton, Franck Prejger, Judith Prihoda, Ian Probert, Gabriele Procaccini, Jeroen Raes, Jeannine Rampal, Josephine Ras, Gilles Reverdin, Emmanuel G. Reynaud, Stephanie Reynaud, Francois Ribalet, Maurizio Ribera d'Alcala, Eric Roettinger, Sarah Romac, Jean-Baptiste Romagnan, Cecile Rottier, Francois Roullier, Christian Rouviere, Anne Royer, Marta Royo Llonch, Martina Sailerova, Guillem Salazar, Gaelle Samson, Sébastien Santini, Christian Sardet, Hugo Sarmento, Eleonora Scalco, Claude Scarpelli, Antoine Sciandra, Sarah Searson, Raffaele Siano, Mike Sieracki, Bianca Silva, Oleg Simakov, Sergei Solonenko, Sabrina Speich, Silvia Spezzaferri, Fabio Stalder, Fabrizio Stefani, Halldor Stefansson, Ernst Stelzer, Lars Stemmann, Lucie Subirana, Matt Sullivan, Shinichi Sunagawa, Jarred Swalwell, Vincent Taillandier, Eric Tambutté, Sylvie Tambutté, Atsuko Tanaka, Isabelle Taupier-Letage, Pierre Testor, Anne Thompson , Doris Thuillier, Virgine Tichanné-Seltzer, Leila Tirichine, Eve Toulza, Sasha Tozzi, Jean-Éric Tremblay, Aline Tribollet, Antoine Triller, Didier Velayoudon, Alaguraj Veluchamy, Emilie Villar, Flora Vincent, Carden Wallace, Markus Weinbauer, Jean Weissenbach, Maureen Williams, Patrick Wincker, Sheree Yau, Alexis Yelton, Adriana Zingone, Didier Zoccola.
The corresponding authors for this work are:
 OCEANOMICS has received funding from the French government, managed by the Agence Nationale de la Recherche, under the grant agreement "Investissement d'Avenir" ANR-11-BTBR-0008.
OCEANOMICS has received funding from the French government, managed by the Agence Nationale de la Recherche, under the grant agreement "Investissement d'Avenir" ANR-11-BTBR-0008.
© 2014 EPEP/CNRS/Station Biologique de Roscoff.